


Cardiopulmonary Imaging
Review
Honeycomb Lung: History and Current Concepts
 Audio Available |
Audio Available |  Share
Share
OBJECTIVE. Originally used to describe the macroscopic appearance of various pathologic processes with multiple cysts, honeycomb lung is now the term used to describe endstage pulmonary fibrosis. The purpose of this article is to discuss the history and imaging of honeycomb lung.
CONCLUSION. Honeycomb lung is considered one of the important CT findings in usual interstitial pneumonia. An additional challenge for radiologists is that different pathologic processes can mimic honeycomb lung. Therefore, a precise understanding of honeycomb lung is necessary.
Keywords: CT, honeycomb lung, idiopathic interstitial pneumonia, interstitial lung disease
| Introduction |
|---|
The term “honeycomb lung” originated in the 19th century in Germany where medicine was highly developed in that era. We can go back as far as 1858 when Kessler described a case of congenital bronchiectasis [1]. Since then, the honeycomb appearance of the lung was named, variously by different authors, Wabenlunge (honeycomb lung), Zystenlunge (cystic lung), Schwammlunge (spongy lung), and Sacklunge (sacculated lung) [1, 2]. In the German literature, honeycomb lung was usually considered a congenital malformation or anomalous development of the lung with bronchiectasis [2] (Figs. 1A and 1B). It seems that the term “honeycomb lung” was not applied to chronic interstitial pneumonia in Germany.
In the first half of the 20th century, the idea of idiopathic pulmonary fibrosis (IPF) was not yet established, and only occasional case reports were found, in which the authors called their cases different names or only reported the clinical and pathologic findings [3–6]. The famous report by Hamman and Rich [3], which initially appeared in 1935, did not use the term “honeycomb lung.” In the subsequent English literature, cases of presumed IPF were reported under the name bronchiolar emphysema [5], pulmonary muscular hyperplasia [4], or cystic pulmonary cirrhosis [6]. Originating in the German literature, these names were translated into English and were discussed as a rare and new disease entity [7, 8]. Sibert and Fisher [5], who used the term “bronchiolar emphysema,” considered their cases to be a special type of pulmonary emphysema.
The term “honeycomb lung” first appeared in the English literature in 1949 in a study by Oswald and Parkinson [9]. Sixteen clinical cases of honeycomb lung were presented in detail. The cases included, as interpreted by current medical knowledge, Langerhans cell histiocytosis, lymphangioleiomyomatosis (LAM), and probable chronic interstitial pneumonia, which was not a disease diagnosed in that era [9] (Figs. 2A and 2B). In the definition by Oswald and Pakinson, honeycomb lung was “descriptive of the appearance of the cut surface of the lungs” as being “thin-walled cysts distributed uniformly throughout the substance of both lungs, varying in sizes up to a maximum of 1 cm in diameter.” A more comprehensive report with a pathologic description followed in 1956 by Heppleston [10], who in his practice dealt with a wide variety of diseases and clinical backgrounds (32 patients were coal workers). In this report, most cases were probable chronic interstitial pneumonia, which also included a small number of those with sarcoidosis, berylliosis, eosinophilic granuloma, LAM, giant cell interstitial pneumonia, scleroderma, and even tuberculous bronchopneumonia.
Thus, before the 1960s, honeycomb lung was considered the macroscopic appearance of lung diseases comprising various histopathologic processes and causes, partly because physicians had little idea of identifying the diseases in the way we do now and partly because of the macroscopic similarity of various cystic lung diseases. It was not until 1965 that honeycomb lung was limited to chronic interstitial pneumonia when Meyer and Liebow [11] suggested honeycombing as “the end-stage of chronic interstitial pneumonia no matter what the etiology.”
 View larger version (137K) | Fig. 1A —57-year-old woman with bronchiectasis. Thin-section CT image shows extensive bronchiectasis involving whole right, lower lobe, simulating honeycomb lung. Note that cysts are lined along bronchial tree. |
 View larger version (105K) | Fig. 1B —57-year-old woman with bronchiectasis. Photograph shows macroscopic appearance of surgically resected right lower lobe. There are multiple thin-walled cysts occupying most of lobe. Note that some cysts contain mucous material (arrows). Appearance was called “Wabenlunge” in Germany in late 19th and early 20th centuries. |
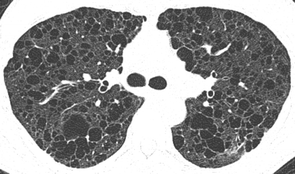 View larger version (114K) | Fig. 2A —24-year-old woman with lymphanigoleiomyomatosis (LAM). Thin-section CT image at level of tracheal carina shows multiple thin-wall cysts, which are mostly discrete and not subpleural, although some cysts are clustered. Appearance is not typical of honeycomb lung by current definition. |
 View larger version (140K) | Fig. 2B —24-year-old woman with lymphanigoleiomyomatosis (LAM). Photomicrograph of surgically resected lung tissue reveals clusters of thin-walled cysts in subpleural area. Fibrosis is limited to area of cyst wall (arrows) and it is obvious that cysts are not result of end-stage pulmonary fibrosis. (Elastica-Goldner stain) |
| Pathologic Description |
|---|
In the initial detailed description of honeycomb lung, Heppleston [10] stated that the essential change is the obliteration of bronchioles by fibrosis or granulomata and compensatory dilatation of neighboring bronchioles, which forms the honeycomb appearance [10]. These changes were the consistent pathologic finding of honeycomb lung, irrespective of the cause.
In another study, honeycomb lung in scleroderma, dermatomyositis, Langerhans cell histiocytosis, tuberculosis, lipoid pneumonia, sarcoidosis, and IPF was studied by continuous sections of three resected lungs and eight autopsy lungs, which were displayed in three dimensions through photographic reconstruction [12]. The authors found that the pathologic process in the honeycomb area was independent of the original disease and was fundamentally diffuse saccular or cystic bronchiolectasis involving the whole lobule, especially the terminal and respiratory bronchioles. The other bronchioles showed abnormal changes in direction, branching pattern, and abrupt amputation, and there were anastomoses between anatomically independent bronchioles. These authors insisted that the primary abnormality of honeycomb lung is the fibrous or granulomatous process of alveoli and ducts, which causes the bronchioles to dilate or amputate.
| Radiologic Description |
|---|
The early radiologic description of honeycomb lung also indicated the various causes and pathophysiology [13]. Honeycomb lung, defined by “visualization of multiple lucent shadows from 2 to 10 mm in size” on a chest radiograph, included not only diffuse interstitial fibrosis but also bronchiectasis and even bullous emphysema [13]. The radiologic concept changed later than the pathologic one. In the 1970s, the concept gradually changed. Reed and Reeder [14] listed cystic bronchiectasis, eosinophilic granuloma, pneumoconiosis, and sarcoidosis in addition to idiopathic pulmonary fibrosis (Hamman-Rich) as the common lung diseases in their gamut of honeycomb lung. However, Felson [15] emphasized interstitial fibrosis as the basis of honeycomb lung and excluded cystic bronchiectasis and bullous emphysema from its definition. Genereux [16] discussed honeycomb lung from the standpoint of the end stage of interstitial lung disease of various causes. This idea coincides with that of Meyer and Liebow [11], who believed that the lung can respond only in a stereotyped manner to a variety of insults. Therefore, various lung diseases can result in end-stage lung (honeycomb lung), which is “characterized by cystic spaces of variable size and extent and is caused by alveolar septal dissolution, bronchiolectasis, and obstructive emphysema.” Significantly, these descriptions were in the era of the chest radiograph, when the macroscopic appearance of the lung was imaged imprecisely and thus had substantial limitations.
 View larger version (134K) | Fig. 3 —62-year-old man with fibrotic nonspecific interstitial pneumonia (NSIP). Thin-section CT image reveals mixture of consolidation and ground-glass opacity with traction bronchiectasis (arrows). These ectatic bronchi simulate appearance of honeycombing. However, they can be tracked back to bronchial tree with accompanying pulmonary artery on contiguous images. Lung tissue intervenes between ectatic bronchi and cystic spaces are not clustered. These features are different from honeycomb lung. |
 View larger version (129K) | Fig. 4A —78-year-old woman with idiopathic pulmonary fibrosis (IPF). Initial thin-section CT image reveals multiple clustered cysts of up to 10 mm in lower lobes. |
 View larger version (125K) | Fig. 4B —78-year-old woman with idiopathic pulmonary fibrosis (IPF). Follow-up thin-section CT image obtained 15 months later shows obvious enlargement of each cyst, largest reaching 20 mm. |
 View larger version (162K) | Fig. 4C —78-year-old woman with idiopathic pulmonary fibrosis (IPF). Photograph of lungs obtained at autopsy performed 2 months after B shows cobblestone appearance of lung surface. |
 View larger version (170K) | Fig. 4D —78-year-old woman with idiopathic pulmonary fibrosis (IPF). Photograph of coronal cut surface of resected lungs shows honeycomb lungs, closely resembling CT appearance. |
 View larger version (144K) | Fig. 4E —78-year-old woman with idiopathic pulmonary fibrosis (IPF). Photomicrograph at area of honeycomb lung shows multiple cysts with different sizes in background of dense fibrous scar. Note presence of bronchial epithelization of some of cysts (arrow). (H and E) |
| Recent Definition of Honeycomb Lung |
|---|
The Fleischner Society has recently revised its glossary terms, in which the pathologic definition of honeycomb lung follows that of Genereux [16] as “destroyed and fibrotic lung tissue containing numerous cystic airspaces with thick fibrous walls, representing the late stage of various lung diseases, with complete loss of acinar architecture” [17]. Katzenstein [18] stated that honeycomb lung “has a characteristic gross appearance, with relatively uniformly sized cysts...that are set in a background of dense scarring” and “microscopically...honeycomb lung is characterized by enlarged airspaces surrounded by fibrosis and lined by bronchiolar or hyperplastic alveolar epithelium. The combination of parenchymal collapse...and collagen deposition accounts for the major manifestation of this lesion.” According to Katzenstein, honeycomb lung is nonspecific as to cause, and a diverse group of diseases can lead to honeycombing (idiopathic interstitial pneumonia [IIP], diffuse alveolar damage, asbestosis, interstitial granulomatous diseases, and eosinophilic granuloma), which corresponds to the previous understanding [11, 18]. In another pathology book, Churg [19] described honeycomb lung as “a variable combination of thick-walled cysts..., which may range from a few millimeters to a few centimeters in diameter, and intervening solid fibrous tissue.” He continued, “In addition to specific diffuse diseases, localized scars of many causes can appear as honeycombed lung,” which indicates a rather nonspecific pathologic condition [19].
 View larger version (136K) | Fig. 5 —45-year-old woman with Langerhans cell histiocytosis. Thin-section CT image shows discrete cystic spaces in center of right upper lobe. Some cysts are thin-walled, whereas others just look emphysematous. Most cysts spare subpleural area and are predominant in upper lobes, which is different from typical honeycomb cyst in usual interstitial pneumonia (UIP). |
 View larger version (165K) | Fig. 6A —68-year-old man with combined emphysema and chronic interstitial pneumonia, who developed squamous cell carcinoma. Thin-section CT image of lung tumor reveals multiple thin-walled cysts and emphysema. Groundglass opacity is noted adjacent to cystic area (arrows), indicating presence of chronic interstitial pneumonia. Cystic spaces are confluent and are larger with thinner walls than those of honeycomb lung. This is considered coexistence of emphysema and chronic interstitial pneumonia and not honeycomb lung. |
 View larger version (111K) | Fig. 6B —68-year-old man with combined emphysema and chronic interstitial pneumonia, who developed squamous cell carcinoma. Photograph of cut surface (corresponding to CT slice) of surgically resected left lower lobe shows multiple cysts of various sizes, giving appearance of honeycombing. |
 View larger version (137K) | Fig. 6C —68-year-old man with combined emphysema and chronic interstitial pneumonia, who developed squamous cell carcinoma. Photomicrograph of cystic space shows destruction and fibrosis of pulmonary parenchyma, with varying-sized cyst, which is separated from normal lung by interlobular septa (asterisks). (H and E) |
 View larger version (125K) | Fig. 6D —68-year-old man with combined emphysema and chronic interstitial pneumonia, who developed squamous cell carcinoma. Photomicrograph shows presence of fibroblastic foci (arrow), which is compatible with interstitial fibrosis. (Elastica-Goldner stain) |
Pathologically, the Fleischner Society definition indicates, “the cysts range in size from a few millimeters to several centimeters in diameter,” so they are macroscopically identifiable [17]. Some researchers use the term “microscopic honeycombing” to describe 1- to 2-mm dilated bronchioles surrounded by airless fibrotic lung on a pathologic specimen [20]. On thin-section CT images, the area corresponds to markedly increased lung attenuation with dilated bronchioles and is therefore different from the macroscopic honeycomb lung. Whether the pathologic process that occurs in the “microscopic honeycombing” has a similar clinical and pathophysiologic significance to that of (macroscopic) honeycomb lung is still to be determined.
A recent definition of honeycomb lung in radiology is based on thin-section CT, which can image the lungs close to their macroscopic appearance [21]. The initial definition by the Fleischner Society, which was published in 1984, stated that honeycomb lung is “a number of closely approximated ring shadows representing air spaces 5–10 mm in diameter with walls 2–3 mm thick...whose occurrence implies end-stage lung” [22], which is in concert with the statement by Meyer and Liebow [11] and Genereux [16]. The second statement by the Fleischner Society, which was published in 1996, said, “Clustered cystic air space, usually of comparable diameters on the order of 0.3–1.0 cm but as much as 2.5 cm, usually is subpleural and characterized by well-defined walls, which are often thick—a CT feature of diffuse pulmonary fibrosis” [23]. These earlier definitions, which were in the era of the chest radiograph in the first version and of CT in the second version, are almost the same except for the size and location. In the latest version from the Fleischner Society, honeycomb lung is defined as “clustered cystic air spaces, typically of comparable diameters on the order of 3–10 mm but occasionally as large as 2.5 cm... usually subpleural and characterized by well-defined walls” [17]. The size and wall thickness of the cysts vary among the authors who use the term. However, “the cystic air spaces of honeycomb lung tend to share walls” [24]. This means that traction bronchiolectasis with intervening lung should not be called honeycombing (Fig. 3). Because of the small size of the cyst, diagnosis should be performed with thin-section CT [25]. Although different disease processes result in the same pathologic and radiologic appearance, recent understanding indicates that “honeycombing is often considered specific for pulmonary fibrosis and is an important criterion in the diagnosis of usual interstitial pneumonia (UIP),” and thus “the term should be used with care [because] it may directly impact patient care” [17].
 View larger version (103K) | Fig. 7A —56-year-old man with emphysema who developed interstitial pneumonia during follow-up. Thin-section CT image shows emphysema in periphery of right lower lobe. There is minimum increase in lung attenuation associated with emphysematous area. |
 View larger version (106K) | Fig. 7B —56-year-old man with emphysema who developed interstitial pneumonia during follow-up. Thin-section CT image obtained 5 years later shows cluster of multiple cysts in area of previous emphysema. This appearance mimics honeycomb lung. However, it is not end-stage of chronic fibrosing interstitial pneumonia but superimposition of groundglass opacity on emphysema. |
The latest definition emphasizes the importance of honeycombing as the diagnostic criterion of UIP. This emphasis originated from the recent consensus statement about idiopathic pulmonary fibrosis published in 2000 by American Thoracic Society and European Respiratory Society, which emphasized the clinical relevance of discriminating UIP from other IIP [25]. Among idiopathic interstitial pneumonia, IPF is a progressive disease, often with acute exacerbation, and shows far worse prognosis than other chronic interstitial pneumonia [26]. Although a new antifibrotic drug has emerged as a possible treatment option for IPF [27], the prognosis is still regarded as poor [28].
Because honeycomb lung is the end stage of interstitial pneumonia, there must be an abnormality that precedes it. In the case of UIP, honeycomb lung is preceded by the presence of patchy ground-glass opacity and reticulation within a secondary lobule [29]. Over time, intralobular reticulation increases and traction bronchiolectasis gradually appears in the area of the ground-glass opacity while the ground-glass opacity diminishes and finally results in honeycombing [29, 30]. The earliest CT abnormality of UIP is poorly understood. In one report of 14 cases of silicosis that developed chronic interstitial pneumonia with honeycombing, the earliest abnormality was faint ground-glass opacity or, less frequently, coarse reticulation [30]. However, this study was performed with 10-mm slice thickness.
The speed of progression of honeycomb lung is poorly understood. In a series of 29 IPF patients, the progression of honeycomb lung ranged from 0% to 11% of the lungs per month (median, 0.4%) and was fastest in the lower lobes [29]. In another series of 14 patients with chronic interstitial pneumonia and silicosis observed for a median of 15.4 years, normal or near-normal lung progressed to honeycomb lung within a median of 12.1 years (range, 3.7–19.1 years) [30]. It is presumed that a long asymptomatic period exists before honeycomb lung develops and the patient becomes symptomatic. With the common use of CT examinations in this era of MDCT, cases of IPF without honeycomb lung are expected to be increasingly found, which challenges radiologists in CT diagnosis.
Although most cases of honeycomb lung are seen in chronic lung disease, it can also occur in a minority of patients with acute interstitial pneumonia and diffuse alveolar damage [31, 32]. This disease is progressive by the day or week, and the result is often fatal. Honeycomb lung is not the initial feature, but it can occur as early as 1 week after the onset of symptoms [31].
The size of the honeycomb cyst usually increases during follow-up [33]. In two autopsy series of patients with honeycombing of UIP, stenosis or abrupt angulations of bronchiole and slitlike structures between the cysts and bronchioles were found, which were regarded as the causes of progressive enlargement of honeycomb cysts [12, 33] (Figs. 4A, 4B, 4C, 4D, and 4E). In another study of 97 patients with end-stage lung disease, patients underwent paired inspiratory and expiratory volume scanning of the lung, and the size of honeycomb cysts was evaluated using multiplanar reformation [34]. The large honeycomb cysts (mean, 20.6 mm; SD, 10.7 mm) tended to have thinner walls and did not communicate with the airways and thus did not change in size during forced exhalation, whereas the small cysts (mean, 2.7 mm; SD, 4.3 mm) did have communication with the airways and their size changed on expiration [34]. The data suggest that the progressive enlargement of honeycomb cysts is partly due to air trapping by the check-valve mechanism.
 View larger version (122K) | Fig. 8A —64-year-old man with silicosis and chronic interstitial pneumonia. Initial CT image shows slight increase in lung attenuation in both lung bases (arrows). |
 View larger version (143K) | Fig. 8B —64-year-old man with silicosis and chronic interstitial pneumonia. In CT image obtained 8 years later, ground-glass opacity is obvious and multiple lung cysts are identified in area of ground-glass opacity, giving appearance of emphysematous cysts (arrows). |
 View larger version (142K) | Fig. 8C —64-year-old man with silicosis and chronic interstitial pneumonia. CT image obtained 5 years after B shows further progression of ground-glass opacity and increased number and size of lung cysts. These cysts are thin-walled and spare subpleural zone, although some cysts are clustered. CT finding is different from honeycomb lung of usual interstitial pneumonia (UIP). |
 View larger version (172K) | Fig. 8D —64-year-old man with silicosis and chronic interstitial pneumonia. Photomicrograph of lung specimen shows multiple lung cysts in area of interstitial fibrosis (arrowheads). Appearance is different from honeycomb lung in UIP. Pathologic diagnosis was also different from typical UIP pattern. Arrows indicate pleural surface. (Elastic-Goldner stain) |
Honeycombing is often considered specific to pulmonary fibrosis and is an important criterion in the diagnosis of UIP [17]. By definition, however, it is not the specific CT finding of UIP, although honeycombing is the common finding of UIP.
In previous literature, honeycombing was identified in 41–100% of UIP, depending on the reported series [35–44]. In one report of 168 cases of suspected IIP, the sensitivity and specificity of the presence of honeycombing in the diagnosis of UIP were 90% and 86%, respectively [45]. Nonspecific interstitial pneumonia (NSIP) and desquamative interstitial pneumonia (DIP), which are the chronic interstitial pneumonias of IIP and are the important differential diagnostic considerations, show honeycombing in 0–30% and 4.3–39%, respectively [37, 38, 46–48]. In acute interstitial pneumonia, the frequency is lower, ranging from 6% to 14% [31, 32, 49].
Honeycomb lung in UIP is usually subpleural and is predominant in posterior and lower lobes [29, 35, 50]. This characteristic distribution distinguishes UIP from other diseases with honeycomb lung. In a study of 61 various end-stage lung diseases (26 UIP, nine sarcoidosis, eight Langerhans cell histiocytosis, five asbestosis, four silicosis, four chronic hypersensitivity pneumonitis, three LAM, one berylliosis, and one talcosis), the characteristic distribution of honeycombing made it possible to diagnose UIP in 88% of the cases, with a high degree of confidence in 67% [51] (Fig. 5).
 View larger version (120K) | Fig. 9A —62-year-old man with emphysema and chronic interstitial pneumonia. Thin-section CT image at lung base shows honeycomb lung. |
 View larger version (153K) | Fig. 9B —62-year-old man with emphysema and chronic interstitial pneumonia. Thin-section CT image at tracheal carina shows paraseptal emphysema with varied thickness of cyst wall. Most anterior cysts appear similar to those of lung base, showing similarity of honeycomb cyst and paraseptal emphysema. |
In addition to the distribution, the extent of honeycomb cyst is another important consideration when diagnosing UIP. It has been reported that the extent of honeycombing ranged from 3% to 21% of the lung parenchyma in UIP [35–44]. On the other hand, the extent of honeycombing in NSIP and DIP is reported as 0.3–3.7% and 0.7–10% of the lung parenchyma, respectively [37, 38, 46–48]. In these reports, both the frequency and extent are significantly higher in UIP than in DIP or NSIP, although there seems to be no threshold that discriminates UIP from other IIP.
| Problems in Diagnosing Honeycomb Lung |
|---|
There are several difficulties in the diagnosis of honeycomb lung. One is the definition of the terminology and the others are associated with radiographic diagnosis with CT. The problem of diagnosis originates from the historical change of the concept and the different circumstances in which it is made. In the previous literature, honeycombing meant nonspecific cystic pulmonary conditions, including bronchiectasis and congenital cystic lung disease. However, in the recent literature, its use is limited mostly to the end-stage lung with interstitial fibrosis.
Other problems affect the CT diagnosis of honeycomb lung in cases with chronic interstitial pneumonia [52]. Although the definition is straightforward, diagnosing honeycomb lung in IPF or UIP is sometimes not easy for radiologists. For instance, in a study of 314 cases of IPF, the interobserver agreement regarding the presence of honeycomb lung ranged from 0.21 to 0.31, which indicates rather poor agreement [36]. This variation could originate from the varied appearance of cystic structures in chronic interstitial pneumonia, in which there can be traction bronchiolectasis, subpleural cysts or bulla, and emphysema, especially of the paraseptal type. Located in the subpleural area, all these structures appear cystic and often have walls. The coexistence of emphysema is reported in the subset of patients with IPF, which is rather a common phenomenon because both diseases are associated with cigarette smoking [53–56]. Although emphysema is pathologically defined as loss of lung parenchyma without fibrosis, fibrosis can coexist in the area with emphysema [57] (Figs. 6A, 6B, 6C, and 6D). Although such fibrosis may be different from UIP [58], the presence of this fibrosis can be a risk factor of acute exacerbation during cytotoxic drug treatment [59] and after pulmonary surgery [60].
Emphysema and cystic spaces can be challenging in the presence of overlapping ground-glass opacity [47]. In a study of 34 patients with either UIP or NSIP and emphysema, emphysematous lung or cystic areas were misdiagnosed as honeycombing in three cases [47]. This situation can occur with pneumonia in emphysematous lung but more problematic is the development of chronic interstitial pneumonia in emphysematous lung during follow-up (Figs. 7A and 7B). NSIP and DIP, which show predominantly groundglass opacity, might show a honeycomb appearance if the abnormality involves the emphysematous area and thus would likely be misdiagnosed as UIP [47].
Furthermore, emphysema and interstitial fibrosis develop and progress simultaneously in the same lung area [61] (Figs. 8A, 8B, 8C, and 8D). The end result is similar to honeycomb lung, which is often relatively thin walled and presents a diagnostic challenge to radiologists.
Finally, paraseptal emphysema, which is one of the types of emphysema, has definite walls, is located subpleurally, and is often clustered (Figs. 9A and 9B). Pathologically, paraseptal emphysema is often accompanied by fibrosis in its walls [62]. There are cases that show paraseptal emphysema in the upper and middle lobes, although there is typical honeycomb lung in the lower lobes. In these cases, these different pathologic processes are often continuous with each other in the subpleural zone. Thus, differentiating the two is almost impossible.
It should be kept in mind that honeycomb lung is the end stage of pulmonary fibrosis. Thus, the development of ground-glass opacity in the area of emphysema should not be called honeycomb lung. However, in cases of the simultaneous progression of fibrosis and airspace enlargement, it might be difficult to reject the term honeycomb lung.
| Future of CT Diagnosis of Honeycomb Lung |
|---|
Because the term “honeycomb lung” is closely linked with the concept of the disease and with the classification of idiopathic interstitial pneumonia, the definition and clinical meaning of honeycomb lung have changed and may change from the present definition in the future. In the meantime, it is important to use the term in consensus, and when it is used, interpreters should have a unanimous opinion and image of what is meant by honeycomb lung.
Disagreement concerning the CT diagnosis of honeycomb lung could partly be explained by the lack of prespecified consensus, and thus reference images, such as the International Labor Office classification of pneumoconiosis, are considered to improve the interobserver agreement [36]. Actually, reference images should be used so that we have a unanimous CT diagnosis of honeycomb lung in diagnosing IPF. Because the final goal is to identify those patients who are likely to show progressive disease and die within several years, the prespecified pattern of honeycombing should be revised on the basis of the results of the prospective observation that will be undertaken, depending on the prespecified consensus. The revision is an urgent task for radiologists considering that the presence and extent of honeycombing is the most important thin-section CT finding in the correct diagnosis of UIP [39–41, 43, 44, 47].
Address correspondence to H. Arakawa ([email protected]).
| References |
|---|


